Differentialdiagnose Primäre Knochentumoren im Sinne einer eigenständigen neoplastischen Erkrankung sind selten. Abgesehen von wesentlich häufigeren Skelettmetastasen kommen im Skelettsystem auch Veränderungen vor, die im Zusammenhang mit endokrinen Erkrankungen stehen und dabei in ihrem klinischen und radiologischen Erscheinungsbild den Eindruck eines primären Knochentumors hervorrufen können.
Endokrinopathien ohne hereditär-syndromalen Hintergrund können skelettale Veränderungen als sekundäre Folge der endokrinen Dysregulation hervorrufen. Sie bestehen entweder in einer generalisierten Osteopathie (zum Beispiel Steroidosteoporose bei Erkrankungen der Zwischenhirns-Hypophysen-Nebennierenachse wie etwa dem Cushing-Syndrom, renale Osteopathie bei chronischer Niereninsuffizienz oder onkogene Osteomalazie bei gesteigerter renaler Phosphatausscheidung) oder sie treten in Form von lokalisierten tumorartigen Veränderungen (Herdbefunden) an einzelnen oder mehreren Knochen oder auch extraskelettal in Erscheinung (zum Beispiel der sogenannte braune Tumor bei Hyperparathyreoidismus oder die sogenannte tumorartige Kalzinose bei chronischer Niereninsuffizienz).
Die Diagnostik solcher Veränderungen setzt ein interdisziplinäres Verständnis für die pathophysiologischen und klinischen Zusammenhänge sowie die morphologischen Befunde (Histologie, Radiologie) voraus, um Fehldiagnosen und therapeutische Fehlentscheidungen zu vermeiden.
Die radiologischen Muster von skelettalen Veränderungen, die vor allem durch einen primären Hyperparathyreoidismus ausgelöst werden können, stehen in ihrer diagnostischen Bedeutung im klinischen Alltag hinter den Laborbefunden zurück.
Eine diagnostische Herausforderung
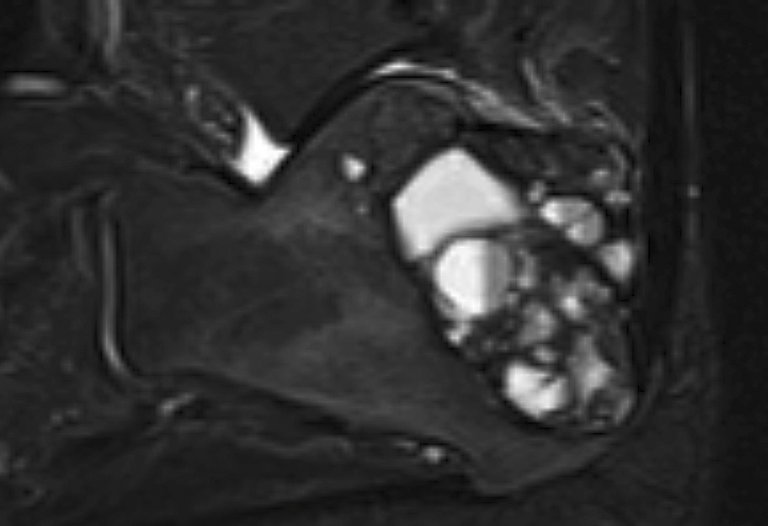
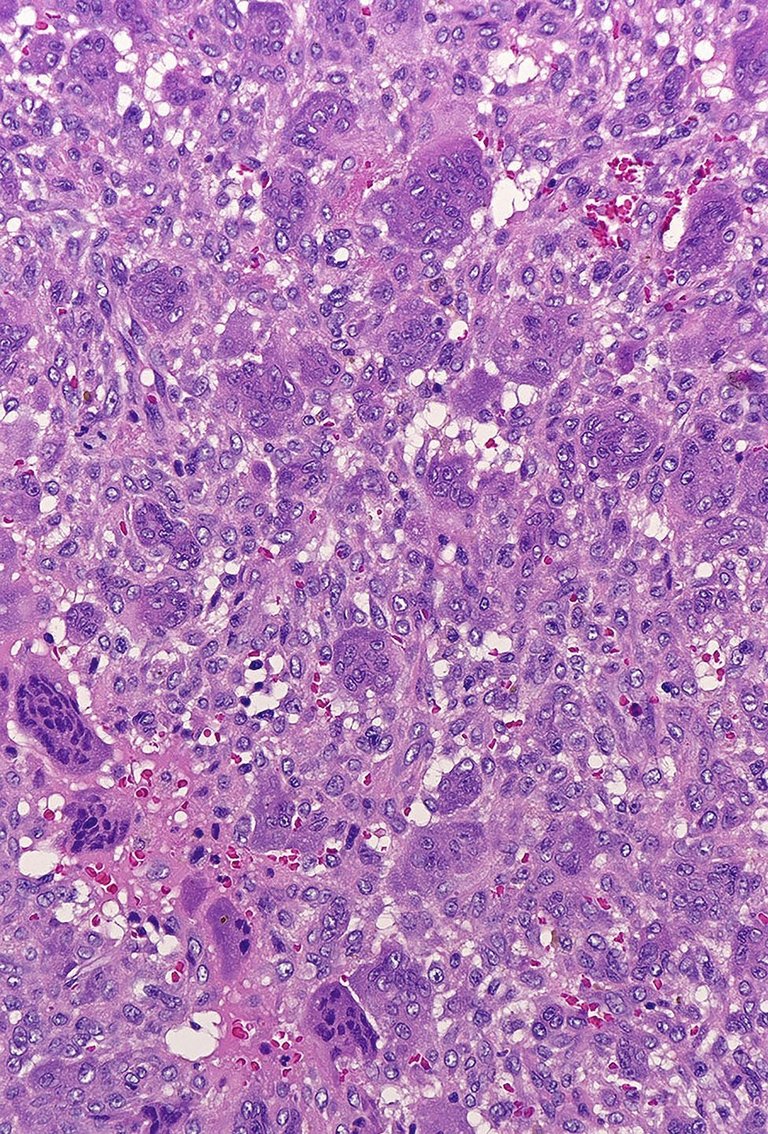
Vorsicht in der differenzialdiagnostischen Abwägung ist geboten, wenn eine Läsion ausserhalb der charakteristischen Vorzugslokalisationen primärer Knochentumoren oder multifokal auftritt. Sehr wichtig ist die sorgfältige Korrelation der klinischen Untersuchungsbefunde und der Laborbefunde des Knochenstoffwechsels zu den radiologischen und histopathologischen Befunden. Auch die mikroskopische Beurteilung einer Gewebeprobe kann zu Fehleinschätzungen und fehlerhaften Therapieentscheidungen führen: Braune Tumoren bei einem Hyperparathyreoidismus können durch ihren hohen Gehalt an osteoklastären Riesenzellen mit anderen riesenzellreichen Läsionen verwechselt werden, wenn der klinische Hintergrund unzureichend berücksichtigt wird (Riesenzelltumor, aneurysmatische Knochenzyste, nichtossifizierendes Fibrom, Langerhanszell-Histiozytose) (siehe Abbildung 1).
Abbildung 1A: Brauner Tumor bei Hyperparathyreoidismus im rechten Calcaneus, multizystische Binnenstruktur ähnlich wie in einer aneurysmatischen Knochenzyste (MRT T2w TIRM, sagittal)
Abbildung 1B: Feingewebliche Aspekt mit einkernigen Zellen und mehrkernigen Riesenzellen ähnlich wie in einem Riesenzelltumor
Die Diagnostik der renalen Osteopathie bei chronischer Niereninsuffizienz und Dialysetherapie erfolgt fast nur noch laborchemisch, so dass die ossären Veränderungen, die durch die Kalzium-, Phosphat- und Vitamin-D-Stoffwechselstörung und im Zusammenhang mit einem sekundären Hyperparathyreoidismus hervorgerufen werden, bei Pathologen kaum noch bekannt sind. Ihre korrekte Beurteilung ist nach der labortechnisch notwendigen Standard-Entkalkung von Knochenproben eigentlich nicht möglich, sondern erfordert eine Einbettung der Knochenbiopsie ohne Entkalkung in Kunststoff und die Anwendung der Hartschnitt-Technik, was bereits bei der Einsendung von Knochenbiopsien mit osteologischer Fragestellung angegeben werden muss. Im Rahmen der Dialysetherapie kann es auch zu tumorähnlichen ossären Veränderungen kommen, die durch die Ablagerung von beta-2-Amyloid hervorgerufen werden (sogenannter Amyloid-Tumor).
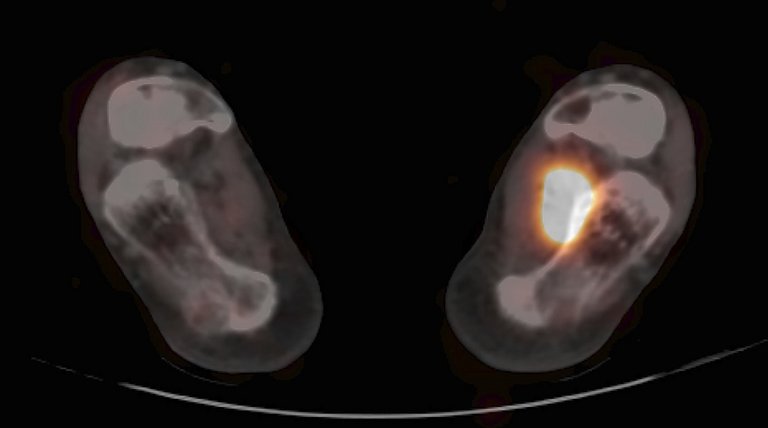
Als Ursache für osteomalazische Veränderungen am Knochengewebe kommen abgesehen vom Vitamin-D-Mangel auch Störungen des Phosphatstoffwechsels in Betracht. Neben den seltenen genetisch bedingten Formen (x-chromosomale, autosomal-dominante beziehungsweise -rezessive hypophosphatämische Rachitis) ist die sogenannte onkogene Osteomalazie zu berücksichtigen, die durch echte Tumoren hervorgerufen wird, die den Fibroblast growth factor 23 (FGF23) sezernieren und dadurch die renale Phosphatausscheidung steigern. Diese Läsionen werden deshalb auch als phosphaturische mesenchymale Tumoren bezeichnet und können im Weichgewebe oder in parenchymatösen Organen lokalisiert sein und sich durch ihre geringe Grösse dem klinischen Nachweis entziehen. Bei Manifestation im Markraum oder an der Kortikalis-oberfläche von Knochen verursachen sie radiologische und auch histologische Befunde, ähnlich wie primäre Knochentumoren, die deshalb früher auch fälschlich als Ursache der onkogenen Osteomalazie betrachtet wurden. Fehldiagnosen sind vermeidbar, wenn eine entsprechende Labordiagnostik zumindest bei unklaren, vermeintlich primären Knochentumoren durchgeführt wird (Serumphosphat, Phosphatausscheidung im Urin, FGF23). Hilfreich für die Lokalisationsdiagnostik kann die Anwendung des DOTATOC-PET/CT sein (siehe Abbildung 2).
Abbildung 2: Phosphaturischer mesenchymaler Tumor in den Weichteilen des linken Fusses (DOTATOC-PET/CT, Fusionsbilder transversal)
Phosphatstoffwechselstörungen kommen in seltenen Fällen auch im Zusammenhang mit der fibrösen Dysplasie vor, insbesondere wenn sie im syndromalem Zusammenhang auftreten (McCune-Albright-Syndrom).
Hereditäre endokrine Syndrome mit skelettalen Veränderungen können am Skelett zu Entwicklungsstörungen, tumorartigen Läsionen oder Tumoren führen. Die ätiologisch jeweils relevanten Gene haben selbst auch Bedeutung im Rahmen der Skelett-Entwicklung oder in der Regulation zellulärer Funktionen von Knochenzellen (Osteoprogenitor-Zellen, Osteoblasten, Chondrozyten). Beispiele dafür sind etwa die kongenitale Tibiapseudarthrose oder die Keilbeinflügeldysplasie bei der Neurofibromatose Typ 1 (NF1), das äusserst seltene Osteochondromyxom bei Carney-Komplex (PRKAR1A) oder das ossifizierende Fibrom im Kieferknochen beim HPT-Jaw-Tumor-Syndrom (HRPT2/CDC73) [1]. Es können auch sekundäre Folgen der endokrinen Dysregulation am Knochengewebe auftreten, wie Hyperparathyreoidismus-assoziierte ossäre Veränderungen beim MEN1-, MEN2- oder MEN4-Syndrom (MEN1, RET bzw. CDKN1B).
Die Diagnostik von skelettalen Veränderungen aufgrund von Endokrinopathien setzt ein interdisziplinäres Verständnis voraus.
Wechselseitige Beziehungen zwischen endokrinen Erkrankungen und Veränderungen am Skelettsystem können zu erheblichen Schwierigkeiten im diagnostischen Alltag führen, deren Bewältigung am besten im interdisziplinären Rahmen gelingt, wie er im deutschsprachigen Raum etwa mit der Arbeitsgemeinschaft Knochentumoren e. V. besteht, in der die beteiligten Fachdisziplinen (Orthopädie, Onkologie, Radiologie, Pathologie) zweimal jährlich Untersuchungsmaterial von diagnostisch interessanten skelettalen Läsionen austauschen und anhand der klinischen, radiologischen und pathologischen Befunde diskutieren (www.agkt.org). Dabei werden auch Fälle aus dem diagnostischen Grenzbereich zwischen primären Knochentumoren im engeren Sinne und endokrin-assoziierten Skelettveränderungen berücksichtigt.
Für Sie zusammengefasst vom:
Jahreskongress DVO: Osteologie 2022
18.-20.09.2022
Baden-Baden
Dr. med. Manoj Kakkassery
Leiter der Muskuloskelettalen Radiologie am Helios Klinikum Berlin-Buch mit dem dortigen interdisziplinären Sarkomzentrum. Er ist spezialisiert auf Knochentumore und Weichgewebssarkome.
Priv.-Doz. Dr. med. Mathias Werner
Oberarzt am Fachbereich Pathologie im Vivantes Netzwerk für Gesundheit Berlin und langjährig spezialisiert auf die histologische Diagnostik von nichtneoplastischen Knochenkrankheiten und Knochentumoren.
Literatur
1 Maraghelli D, Giusti F, Marini F, et al. Bone tissue and mineral metabolism in hereditary endocrine tumors: clinical manifestations and genetic bases. Orphanet J. Rare. Dis. 2020;15:102.