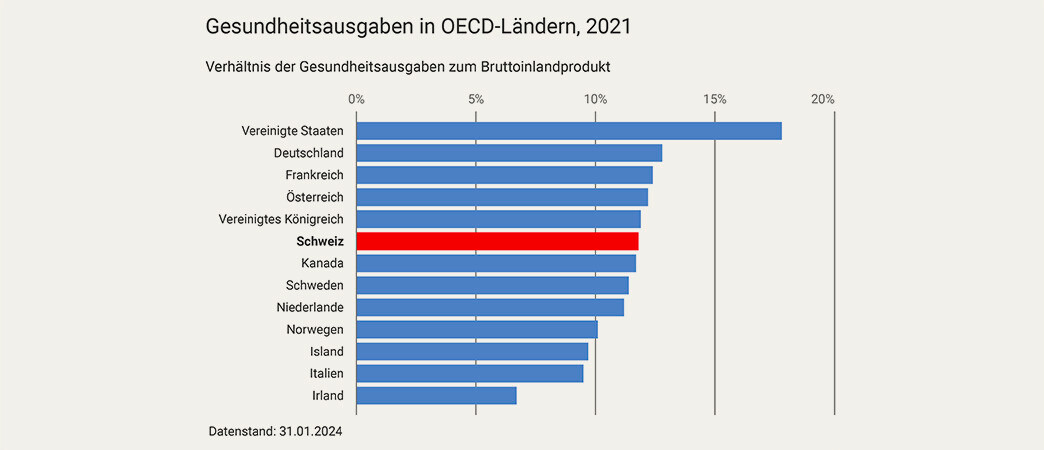
Die Kostenbremse-Initiative würde die Rationierung der Versorgung in der Verfassung verankern und sich auf unsere Lebenserwartung auswirken. Ein im Jahr 2000 in Kraft getretener Ausgabenplafond hätte die im Jahr 2023 erbrachten Leistungen um ein Drittel verringert. TARDOC muss endlich genehmigt werden, um die Zugänglichkeit und Qualität unseres Gesundheitssystems zu sichern.